
Let us make an in-depth study of the laboratory techniques used in microbiology. The serology is grouped into two types: 1. Agglutination (in which particulate antigen is involved) and 2. Precipitation (in which soluble antigen is involved).
Contents
- Laboratory Techniques:
- The Use and Care of the Microscope:
- Common Laboratory Media:
- Preparation of a Smear for Staining:
- Gram’s Stain:
- Ziehl-Neelsen (Zn) Stain:
- Serology:
- Recent Parasitic Serology:
- Cultivation of Animal Viruses:
- Cultivation of Fungi:
- Study of Blood Cell (Hematology):
- Stool Examination for Parasites:
- Examination of Skin Scrapings for Identification of Mites:
- Examination of Urine:
- Application to Nursing:
Laboratory Techniques:
Introduction:
The intelligent use of laboratory services is necessary for the practice of good clinical medicine. The help of the microbiological investigations is quite essential for the correct diagnosis of many diseases and the selection of a suitable antibiotic for the treatment of the patient; without the knowledge of in vitro antibiotic sensitivity of the causative microorganisms, it may not be possible to treat accurately.
Before undertaking in vitro antibiotic sensitivity test of the causal agents of the diseases, it is of great necessity to isolate these agents in pure culture for their identification by the various morphological characteristics after staining them with the appropriate stains and by their biochemical properties.
Parts of Microscope:
Simple magnifying glasses were in use in ancient times, but, later, the combinations of two or more lenses arranged in a tube — so that one lens magnifies the image produced by another — is used, as in the compound microscope. This kind of microscope was originally developed in 1602-1680 by a Jesuit scholar, Athanasuis Kircher.
The lens nearest to the object is known as the objective. The powerful microscope used in the microbiological laboratory has a very small objective lens with a very short focal distance. This lens should be very close to the object. The second lens, magnifying the image made by the objective, is some inches (cms.) above to the first lens.
The eye is applied to the second lens which is called as eyepiece or ocular. Modern microscope has added accessory lenses — both objective and ocular—to make the image very clear and free from distortion, but the basic principles remain the same. The highest magnification given by an ordinary objective is about 90 x.
This image is magnified by a 10 x ocular, giving a total enlargement of the image of 900 x. A good microscope with several combinations of object and ocular, giving total magnification of varying from 50 – 2,000 x are now available. A strong source of light is provided by a mirror by reflecting the light upwards through the object.
A sub-stage condenser has a system of lenses, between the mirror and the object, which collects the light rays and brings them to focus on the objects so that the object is well-illuminated. An iris diaphragm is also included with the condenser to adjust the light.
The object mounted on a glass slide is placed on the flat metal plate/stage, which is between the condenser and the object. To correct the local distance, the wheels are raised or lowered so that the object is focused. Large wheel for coarse adjustment and small wheel for fine adjustments are used. A mechanical stage can hold the slide in position and move the slide to various directions.
Oil Immersion Lens:
The object lens commonly used in the microbiology laboratory to study the bacteria, is an oil-immersion objective (100 x) Fig. 62.1.


 The Use and Care of the Microscope:
The Use and Care of the Microscope:


Objectives:
1. To introduce the student to the effectiveness of the microscope as a laboratory tool.
2. To demonstrate the correct ways of using a microscope.
3. To help the student to understand the precautions that must be taken to protect the microscope from damage.
Electron microscopy can be used to study the extremely minute objects and the inner, minute structure of bacteria and viruses. It makes possible magnification up to 300,000 diameters. Visible light consists of spectrum of electromagnetic waves with wave lengths ranging from about 4000 Angstrom units (Violet) to about 77,000 A° (Red), — A° = 1,000,000 mm. or 0.001 p. Angstrom was a famous Swedish physicist.
Objects smaller than bacteria do not reflect such long waves to the eye in a clear pattern (i.e., are not resolved) and are, therefore, not clearly visible. Electrons create electromagnetic waves of very much shorter length (about 0.05 A0) and when focused magnetically as in electron microscope are capable of resolving objects much smaller than even the smallest bacteria of rickettsiae, particularly viruses and their internal structures.
Electron waves are not visible to the human eye just as ultraviolet rays. The magnified image produced by electron beam in an electron microscope must, therefore, be viewed on a fluorescent screen similar to that used in clinical X-ray fluoroscopy, photographs of the images can be enlarged and are called as electronographs. There are other types of microscopes.
Common Laboratory Media:
For rapid multiplication of bacteria in the laboratory, following growth requirements must be provided:
(a) Food — Most pathogenic bacteria require food as source of organic carbon and nitrogen;
(b) Moisture—Since bacterial cell consists of water, moisture is essential for its growth;
(c) Temperature — For each species, there is a maximum and minimum temperature at which the growth is possible. The optimum temperature for most pathogenic bacteria is 37°C;
(d) Reaction — Like temperature, there is an optimum pH for each species. Usually most pathogens grow well at neutral or slightly alkaline pH (7.2);
(e) Oxygen — Certain bacteria grow in presence of molecular oxygen and are called as ‘aerobes’; others, called as “anaerobes” will grow only in absence of oxygen.
Preparation of Culture Media:
In the laboratory, bacteria can grow on “culture media” which are mixtures of suitable food materials required for bacterial growth. After preparation the pH of the media is adjusted by pH meter or comparator (Fig. 62.2) to 7.2 to 7.4. Prepared media are sterilised by autoclave and poured in Petri dish (plate), test-tubes, flasks etc.
They are incubated to check the sterility and finally they are stored in the refrigerator. For general application, the media are prepared from the base — peptone water (containing peptone, sodium chloride, water) — to which meat extract, blood, serum has been added. For solidifying the media, the agar or gelatin is added.


1. Peptone Water:
Contains 1% peptone and 0.5% sodium chloride dissolved in water. It does not contain meat extract. The medium is colourless. Peptone water is used to study proteolytic activity of bacteria, viz; formation of indole and hydrogen sulphide (H2S.) Motility of bacteria is best studied from peptone water culture; peptone water is used as a base for sugar media.
2. Nutrient Broth:
It is a pale yellow coloured liquid. The first step is to prepare the meat extract. This is done by extracting meat in cold with water. It is then heated to coagulate the dissolved proteins and filtered. The filtrate is the required meat extract. Peptone and sodium chloride are added to the above for preparation of nutrient broth. It is sterilised by autoclaving. It is a common fluid medium for non- fastidious organisms and serves as a base for preparation of solid media.
3. Glucose Broth is a pale yellow liquid which contains basically nutrient broth to which one per cent glucose is added. It is mainly used to grow anaerobic streptococci.
4. Sugar Media:
Various carbohydrates, alcohols etc. (designated as ‘SUGAR’) are fermented by bacteria with the formation of acid or acid and gas. Sugar media are used to study these fermentation reactions. Appropriate sugars are added to peptone water in the proportion of one percent.
A suitable indicator is also incorporated to detect the formation of acid. Usually Andrade’s indicator is used, which is colourless in alkalipe and neutral pH and turns pink in presence of acid. To detect the production of gas, a small tube (Durham tube) is placed in an inverted position in the medium. The air is replaced by fluid during sterilisation by heat.
Gas, if produced, is seen as a bubble at the top of the Durham tube. The sugar media are sterilised in a steam sterilizer at 100°C for 20 minutes on THREE SUCCESSIVE DAYS. The common sugar media are Glucose, Sucrose, Lactose, Mannite, and Dulcitol. Note the coloured plugs to differentiate them (Glucose; Blue; Sucrose White. Mannite, Pink; Lactose, red; Ducitol, Yellow).
5. Urea Medium:
It contains peptone, sodium chloride, glucose, buffer, 20% urea, phenol red, pH 6.8 – 6.9. After inoculation heavily with cultures and incubation at 37°C for 4 hrs, and then overnight, urease positive cultures produce purple pink colour due to a change in the colour of the indicator. Thus, urease producing organisms are identified.
6. Glycerol Saline Transport Medium:
This medium contains glycerol, sodium chloride, buffer, phenol red, water. It is used to transport enteric bacilli to the laboratory. This fluid should not be used if it becomes acid, indicated by a change in colour to yellow.
7. Cary Blair Medium:
This is a buffered solution of sodium chloride, sodium thioglycollate, disodium phosphate and calcium chloride at 8.4. It is a suitable transport medium for Vibrio cholerae. At pH 7.4, it can be used to transport Salmonella, Shigella and Campylobacter.
8. Stuart’s transport medium is composed of anaerobic salt solution, methylene blue, agar 0.6% pH 7.3 – 7.4.This is soft agar medium used to maintain the viability of gonococci on swabs during transport to the laboratory.
9. Venkatraman-Ramakrishnan (VR) Medium:
A simple modified form of this medium is prepared by dissolving 20 g. Crude sea salt and 5 g. peptone in one liter of distilled water and adjusting the pH to 8.6 – 8.8. It is dispensed in screw-capped bottles in 10 – 15 ml amount. About 1 – 3 ml stool is to be added to each bottle. In this medium Vibrio Cholerae do not multiply, but remain viable for several weeks.
1. Nutrient Agar is prepared by adding agar to nutrient broth in proportion of 2 to 3 percent. The broth is put in the steamer to dissolve the agar. The agar broth fluid is filtered hot if necessary and autoclaved. It becomes solid when cooled. The medium is almost transparent. It is a common — purpose solid medium for growth of non-fastidious organisms, further it serves as a base for the ‘Enriched media'(Fig. 62.3).

2. Blood Agar is prepared by adding defibrinated or citrated blood to nutrient agar in the proportion of 5 to 10 per cent. Blood is collected aseptically and added to the already sterilised nutrient agar which has been melted and cooled to about 55°C. The medium is used for the cultivation of fastidious organisms which fail to grow on nutrient agar.
3. Chocolate Agar is prepared by adding, the defibrinated blood, heated to 55°C, in the proportion of 5 to 10 per cent, to sterilised nutrient agar which has been melted and cooled to 55°C. It is opaque brown in appearance. This medium is used for the cultivation of gonococci and meningococci which form greyish colonies.
4. Tellurite Chocolate Agar: It is heated blood agar to which potassium tellurite (0.04%) has been added. It inhibits the growth of many commensal organisms likely to be present in the throat but not the diphtheria bacilli and related organisms. Moreover these latter organisms form grey or black colonies on the medium as they reduce potassium tellurite. The medium is opaque brown in appearance.
5. Loeffler’s Serum: Consists of human or animal serum added to Glucose broth. The collection of blood and separation of serum are carried out with aseptic precautions. The medium is solidified by inspissation. The medium is opaque white in appearance. It is mostly used for cultivation of the diphtheria bacillus. It can be employed to study proteolytic activity of bacteria (liquefaction of serum).
6. Dorset’s Egg Medium is prepared by inspissating a mixture of whole egg fluid 3 part and saline 1 part. The medium is yellow and opaque. When glycerinated, the medium is used for cultivation of tubercle bacilli; without glycerine it is useful for the preservation of stock cultures of enteric bacilli.
7. Lowenstein Jensen Medium: It contains a number of mineral salt, glycerine, starch, egg proteins and malachite green. The last is a bactericidal agent. This medium is used for the cultivation of tubercle bacilli.
8. MacConkey Medium. (Bile salt lactose Neutral red agar): It is used for the isolation of intestinal pathogens. Bile salt inhibits the growth of Gram-positive cocci. Pathogenic Gram-negative enteric bacilli (Salmonella, Shigella) do not ferment lactose and form pale colonies. Escherichia coli (which are mostly commensels), on the other hand, ferment lactose producing acid and, therefore, their colonies are pink. The medium is thus both an inhibitory and differentiating one.
A number of media are described which work on the same principles, viz., D.E.C. medium of Punjab and Ghosh and Leifson’s desoxycholatecitrate medium etc. They differ from the basic medium described above in containing inhibitory substances other than bile salt. All appear reddish brown and transparent (Fig. 62.4).
9. Wilson and Blair Medium: This medium is utilised for isolation of typhoid and paratyphoid organisms from heavily contaminated material like sewage. It contains sodium sulphate, Bismuth ammonium citrate and brilliant green. The last even inhibits Escherichia coli. Typhoid and paratyphoid organisms form black colonies as they reduce to sulphate to sulphide in presence of glucose precipitating insoluble bismuth sulphide (which is black). It is also a differentiating and inhibitory medium.
10. Robertson’s Cooked Meat Medium: Minced meat is boiled in alkaline water. Softened meat is then partially dried between folds of filter paper and added to nutrient broth. The medium is autoclaved for sterilisation. The medium is used for cultivation of anaerobic organisms.
11. Litmus Milk Medium is prepared by adding litmus solution to skimmed milk. The medium is autoclaved or boiled for sterilisation. It is used for studying proteolytic and saccharolytic activity of Clostridium welchii.
12. Thiosulphate Citrate Bile Salt Sucrose (TCBS) Medium: This medium containing thiosulphate, citrate, bile salt, bromothymol blue and sucrose is available commercially and is very widely used at present. Vibrio Cholerae produce large yellow convex colonies which may become green on continued incubation.
13. Triple Sugar Iron (TSI): This medium contains glucose, lactose and sucrose, phenol red indicator (to indicate fermentation), ferrous sulphate (to demonstrate H2S, indicated by blackening of the butt). It is orange red when un-inoculated. It is used to identify the enter obacteriacae. The glucose concentration is 1 /10th of concentration of lactose and sucrose so to detect the fermentation of glucose alone.
The small amount of acid produced by fermentation of glucose is oxidised rapidly in the slant which reverts to alkaline (red); in contrast, under lower O2 tension in the butt, the acid reaction (yellow) is maintained. Free exchange of air may be permitted in the slant through the use of loose cotton plug to promote oxidation of acid produced by glucose fermentation in the slant. This reaction should be read after 24 hrs. incubation (Fig. 62.5).


14. Simmons’ Citrate medium is modified Koser’s medium with agar and an indicator (bromothymol blue). It is used to test the ability of an organism to utilise citrate as the sole source of carbon and energy for growth and an ammonium salt as the sole source of nitrogen. It is also called as citrate utilisation test.
Result:
Positive = Blue colour and growth of bacteria.
Negative=Original green colour and no growth.
Both Wilson Blair and Lowenstein Jensen media are green. Wilson Blair, however, is usually poured in Petri dishes whereas Lowenstein Jensen medium has to be kept in screw capped bottles or tubes with paraffinised plugs.
Agar Agar:
It is a polysaccharide prepared from a sea-weed grown on the Japanese and Chinese sea coasts. It is available in dry strands or in powder forms. It has no nutrient property and is used for solidifying the media. It does not set till it is cooled to 42°C. Therefore, blood, serum etc. can be added to nutrient agar at 55°C at which temperature the medium is still fluid and there is no risk of denaturation of the serum proteins added.
Inoculation of Culture Media:
In clinical specimens (feces, sputum etc.), mixtures of microorganisms are very common. It is a mixed culture. In the microbiological laboratory, to study a causative agent of the particular disease, it is necessary to inoculate the specimens into the media and fish out a single bacterium from the pure culture for its further identification in the laboratory.
There are various methods of isolation of bacteria in pure culture. Of which, the plating or streaking (inoculation of culture media) is the most common easy method —”Specimens or materials containing bacteria should be streaked several times over the surface of a solid medium with a platinum loop without recharging it; fewer and fewer bacterial cells Will remain on the loop, as the successive streaks are made and finally it may deposit a single bacterial cell on the surface of the media. Thus separate colonies are obtained.”
Preparation of a Smear for Staining:
This is the first method in the staining of bacteria. Smears are made on a clean, grease-free, dry slide. For examining a culture on a solid medium, the required colony is touched with the tip of a platinum loop or wire and a small loopful of the material thus removed is mixed with a drop of water or normal saline to give a faint turbidity.
Then the drop is spread into a thin film and allowed to dry in air or by gently warming over the flame. If the culture for examination be in a fluid medium, a drop of the medium is removed with the platinum loop and a film is prepared from it directly. Similarly, smears are prepared out of any fluid material like sputum, pus etc., collected from a patient.
At last, the smears are “fixed” by passing them through a flame four to five times. The fixation kills the bacteria and — by coagulating any protein contained in the preparations, makes the bacteria to adhere firmly to the slide (Fig. 62.6).
Gram’s Stain:
1. The fixed smear is covered with Oxalated gentian violet; allow the stain to act for one minute.
2. Pour off excess of the stain, holding the slide at an angle downwards, wash away the remain of the stain with water and add Gram’s Iodine. Allow Gram’s Iodine to act for THREE minutes.
3. Wash off Iodine with Ethyl alcohol, and continue with fresh alcohol drop by drop, until no more violet colour is removed from the film. This step is called decolourisation.
4. Wash with tap water.
5. Counterstain with safranin for ONE minute. Safranin stains only those bacteria which have completely decolorized by alcohol.
6. Wash with tap water and dry the smear between blotting paper. See that the smear is completely dry.
7. Examine under oil immersion lens (Fig. 62.7).

Result:
With Gram’s bacteria can be divided into two large groups:
1. Gram-positive; and
2. Gram-negative.
Gram positive bacteria retain gentian violet stain despite decolourisation and stained violet. Gram-negative bacteria loose completely the violet stain after decolourisation with alcohol and stain red by the counterstain, Safranin.

Ziehl-Neelsen (Zn) Stain:
Cell wall of certain bacteria (i.e., Mycobacterium tuberculosis) contains wax like substance known as “mycolic acid’.’Hence, these bacteria cannot be easily stained by Gram’s stain. Heat or prolonged contact is required to drive the stain into the bacterial cells; once stained, they are not decolorized by mineral acids. They are called “acid fast bacilli” (AFB).
So Ziehl-Neelsen stain has been devised to study their morphology microscopically:
1. Cover the fixed smear with carbol fuchsin. Now heat the smear gently from below till steam rises.
2. Allow the stain to act for five minutes and, during this time, steam twice again.The stain must not be allowed to evaporate and dry on the smear.
3. Wash the smear well with tap water for TWO minutes.
4. Pour on the slide 20% H2SO4 from drop bottle. The red colour of the smear becomes yellowish brown. After ONE minute wash with tap water. If the red colour returns, pour acid again. The decolourisation is continued till, after washing, the film is only faint pink.
5. Wash well with tap water TWO minutes.
6. Counterstain with Methylene blue for 30 seconds.
7. Wash with tap water ONE minute and blot and dry.
8. Examine with oil immersion lens (Fig. 62.8).

Thus with Ziehl-Neelsen stain, acid fast bacilli (tubercle bacilli) are stained pink. Non-acid fast bacilli lose the pink colour by treatment with mineral acid and take the counterstain. They are stained blue.
Hanging Drop Method:
(For the study of motility of bacteria):
Method:
Take a “hollow ground glass slide” and cover the surface around the hallow with a thin layer of vaseline. By means of platinum loop which has already been sterilised by flaming, place a drop of bacterial suspension in the centre of a clean cover glass.
The slide is then gently lowered on the cover glass so that the drop remains undisturbed in the hollow. The slide is then turned over and the drop is first examined with the low power objective to locate the edge of the drop. Then the motility should be observed by high power objective.
The vaseline fixes the cover glass and tends to prevent the evaporation of the bacterial drop during prolonged observation. The examination should be performed with the light partially cut off, i.e., with the iris diaphragm of the microscope appropriately closed, after the condenser already been lowered down (Fig. 62.9).


Antibiotic Sensitivity Test:
Subcultures of the test organisms are made in broth and incubated for 6 -18 hours. Any density of inoculum may be used but, in most cases, the surfaces of cultures plates may be sown with broth containing approximately 100 million cells per ml. After drying the surface, the previously prepared disks of filter paper saturated with various concentrations of antibiotics are applied, the plates are incubated and the zone of inhibition is measured.
A zone of inhibition of growth around the disk may indicate that the antimicrobial agent is either bactericidal. (S = bacteria are sensitive) or merely bacteriostatic (R R = bacteria are relatively resistant) or bacteria are resistant (R = resistant). If the test organism is not resistant to the antibiotic, zones of complete inhibition of the growing organism are found around the disks containing the effective antibiotic (Fig.62.10).

Culture and sensitivity test report of diagnostic microbiological laboratory.
Culture-organism isolated. Staphylococcus aureus
Sensitivity:
Penicillin — R
Tetracycline — S
Gentamycin — S
Streptomycin — RR
(R = resistant; S = sensitive; RR = relatively resistant).
Serology:
Serology is the study of reaction between antigen and antibody and is grouped broadly as follows:
1. Agglutination (in which particulate antigen is involved); and
2. Precipitation (in which soluble antigen is involved).


1. Agglutination:
Serological techniques are of considerable importance in identifying unknown antigens and detecting the presence of antibody in the serum of infected or immunized subjects. Agglutination tests are performed by mixing a serial dilution of antiserum with a suspension of bacterial cells or particles. These methods are used to examine sera for antibodies against Salmonella and Brucella and, using standard antisera to identify the unknown organisms.
The basic principle of the techniques of agglutination tests in enteric fever (typhoid or paratyphoid) is to examine the serum quantitatively for agglutinin towards a particular organism. For this purpose, the method usually adopted is to mix varying dilutions of serum (made up in saline solution) in narrow tubes with a fixed quantity of a uniform and stable suspension the organism or its antigen.
These mixtures are kept at 37°C or 50 – 55°C in a water bath for a certain length of time and then examined for visible agglutination of the suspension. The agglutinated antigens tend to sediment and the reaction can also be evaluated by the amount of deposit in the tubes and the clarity of the supernatant fluid. The strength can be stated in terms of the highest dilutions (titers) that produce the agglutination.
The measuring titer:
The titer of an antiserum is the number of antibody units per unit volume of the original serum; thus if the last tube, showing a reaction, e.g., agglutination, contains 1 ml volume and is a dilution of 1 :128 of the original serum, the titer of the serum is 128 units of antibody per ml of the serum.
At least, 5 ml of blood should be obtained by vein puncture and the blood is immediately transferred from the syringe to a dry well-stoppered sterile bottle or tube and allowed to clot; when the serum has separated, it is pipetted off into a sterile tube.

(a) Widal Test:
In the routine laboratory diagnostic Widal reaction, the patient’s serum is tested simultaneously with the antigen of each organism likely to be responsible for enteric fever in the particular region, e.g., Salmonella typhi or S. paratyphi A or B or C. Additional information can be obtained by testing separately for H and O agglutinins. Thus, Widal test generally involves parallel tests with different Salmonella group of organisms and also different forms of the same organism.
In addition to the test with typhoid-paratyphoid organisms, it is the practice of many laboratories to test also for Brucella melitensis agglutinins and, if considered necessary, with non-motile strains of proteusox19,ox2and oxk for typhus fever, thus increasing the number of parallel tests. This is called as ‘Triple Widal’:
Master (1:15) dilution of the patient’s serum is first prepared, thereafter a series of doubling dilutions are made in narrow tubes. Then, 0.4 ml of the bacterial antigen is added in each tube. To observe H type (loose cotton wool) agglutination, it is usually sufficient to incubate at 37°C for 2 hours and leave for half an hour at room temperature/large flake “clumping or agglutination can easily be detected with naked eye. The flocculi can also sediment rapidly and the deposit is quite perceptible in the narrow tube (Dreyer’s tube).
Compact agglutination of O type settles down in round bottom tubes (Felix tubes) kept in the first row of test tube rack and its readings should be made after 4 and 24 hours of incubation as these forms of reactions develop slowly. The control tube contains saline only. The clumps are small, granular, and visible with the help of hand lens. O agglutination is always positive as O antigen is common to all Salmonella. If 0 and H agglutinations are positive, then it is diagnosed as Typhoid since the causative agent, S. typhicontains both O and H antigens.
If AH agglutination is positive, then it is considered as paratyphoid caused by S. paratyphi A. Widal test report of diagnostic laboratory.
I. For Typhoid

II. For Paratyphoid:
S. typhi O — 1:60 S. typhiH — Negative
S. paratyphi A— 1:120 Paratyphoid by S. paratyphi A – positive
S. paratyphi B — Negative
S. paratyphi C — Negative (Fig. 62.11 a, b)


The same agglutination techniques of Widal test are also applicable to diagnose brucellosis, typhus fever in man. A titer of 1:40 or 80 International Units (IU) per ml. is considered positive for Brucellosis (c) and a titer of 200 or a peak titer of 1,000 – 5,000 is positive for Typhus fever (b).
(b) Cold Agglutination Test:
In cases of primary atypical pneumonia caused by Mycoplasma hominis, the serum may agglutinate at low temperature group O human red blood cells. To a series of doubling dilutions of patient’s serum (1 :8 to 1 :2,048) an equal volume of 0.2 percent of suspension of washed group O human erythrocytes is added. The mixtures are kept in the refrigerator at 0° – 4°C overnight. Agglutinations are observed with naked eye. A titer of 1: 32 to 1: 64 is considered significant.
(c) Paui-Bunnel Test:
An agglutinin for sheep red blood cells is present in the serum and is of diagnostic significance during and after the attack of infectious mononucleosis.
Test. The patient’s serum is heated at 55°C for 20 minutes and series of doubling dilutions of this serum is made from 1:16 to 1:1,024.The control tube contains saline only. 0.5 ml. of a 1 per cent suspension of saline washed sheep red blood cells is added to each tube containing diluted serum. The tubes were thoroughly shaken and incubated at 37°C for four hours.Tube showing agglutination of red blood cells is the final dilution of the serum. A suggestive titre is 1:128; a significant titre is 1:256.
(d) Rose Waaler Test or Test for Rheumatoid Arthritis or Serum Factor:
Sheep red blood cells sensitized with rabbit anti- sheep erythrocytes serum are agglutinated by the sera of 70 – 80 per cent patients with rheumatoid arthritis and positive reaction occurs in some 2 – 5 per cent of normal subjects. Serum factor is macroglobulin which is an index of inherited metabolic disturbances which predisposes an individual to rheumatoid arthritis and is an antibody to denatured gamma globulin.
Previously inactivated patient serum is diluted in two fold steps from 1:2 to 1:1,024. Two sets of dilutions are required; to one set is added an equal volume of sensitized red blood cells and the second set receives the same volume of unsensitised red blood cells of sheep and acts as control.
The tubes are incubated at 37°C and then placed in the refrigerator until the cells have completely settled. The last tube or last wall of Perspex plastic plate, if used, shows a definite hemagglutination at the end titre. Serum titre of 16 or more is taken as positive in rheumatoid arthritis.
(e) Latex Agglutination Test for Rheumatoid Arthritis:
To one drop of diluted patient’s serum on the latex plate, one drop of latex antigen is added and mixed thoroughly with the plastic stick. Latex agglutination indicates that the test is positive.
(f) Coomb’s Test:
Mechanism. In Coomb’s anti-globulin test, the red blood cells (RBCs) anti-D antibody (IgG) cannot linktwored cells for aggluination. Addition of Coombs’ serum (anti-globulin, AHG) has brought about agglutination by linking two attached immunoglobulin’s to one another. It may be direct or indirect Coombs’ test. Both these tests can detect incomplete antibodies.
(A) Direct Coombs’ test detects autoimmune hemolytic anemia, erythroblastosis fetahs.
Method:
To remove serum proteins except globulin, RBCs are washed three times in saline by centrifugation. When washed RBCs are mixed with AHG in presence of bovine albumin, there will be agglutination of RBCs in positive cases.
(B) Indirect Coombs’ test can detect anti Rh antibody in serum of Rh negative pregnant women of Rh positive husband.
Method:
Saline washed Rh positive group 0 red cells are mixed with patient’s serum and incubated at 37°C for 30 minutes. Red cells are washed three times in saline. These washed cells are mixed with Coombs’ serum. Clumping of RBC soccufs, if positive.
2. Precipitation:
(a) VDRL Test:
Venereal Disease Research Laboratory (VDRL) flocculation test is simple, rapid, sensitive screening test used to diagnose syphilis. One drop (0.05 ml) of inactivated patient’s serum is placed on a VDRL glass plate and one drop of diluted VDRL antigen Cardioli- pin, 0.03 per cent; Lecithin, 0.24 per cent and cholesterol, 0.9 per cent), is added on the serum and the whole plate is rotated on VDRL rotator for 3 to 4 minutes, visible aggregation or precipitation indicates a positive reaction.
(b) Kahn flocculation Test to diagnose syphilis. The test for each serum is set up as follows:
The tubes are shaken in a Kahn shaker machine for 3 minutes and incubated at 37°C for 20 minutes. Then add saline 1.0 ml. 0.5 ml. 0.5 ml.
Readings are now made after the appearance of precipitates.
The control is set up as follows:
Reading:++++ = large floccules at the bottom of the Kahn tube; – = minute floccules just visible throughout the tube.
(c) Complement Fixation Test (CFT):
Complement fixation test (CFT) is frequently employed with soluble antigen for the detection of antibody, even 0.08 ^g of antibody nitrogen can be detected. It is very sensitive and specific. Both bacterial and viral infections can be diagnosed with this test.
When antigen and antibody reaction is specific, the complement is fixed. This reaction cannot be seen with the naked eye; but it can be observed with the help of hemolytic system (Sheep RBCs + Hemolysin); if the complement is fixed, there is no hemolysis (Positive); if the complement is free, there is hemolysis (test is negative).
When CFT is employed for the diagnosis of syphilis, it is called Wassermann test:
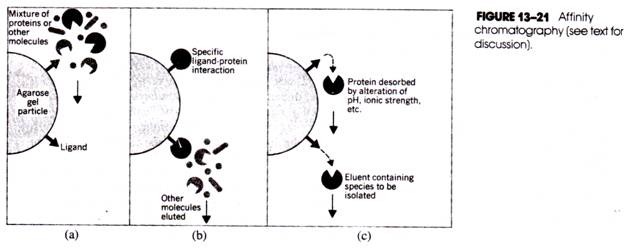
(d) Agar Gel Precipitation Technique:
In this test, the antigen and antibody diffuse is the gel and form a line of precipitation when they meet. When this basic principle is adopted in electrophoresis apparatus, it is known as Counter Immunoelectrophoresis (CIEP).
(e) Fluorescent Antibody Technique:
There are two main methods used in immuno-fluorescent work: the direct and indirect methods. The direct method consists of bringing fluorescein tagged antibodies in contact with antigens fixed on a slide, allowing them to react, washing off the excess of antibody and examining under the fluorescence microscope. The site of union of the labelled antibody with the antigen can be determined by the apple green or orange yellow fluorescent area on the slide.
The indirect method is used to detect both specific antibody in sera and also, as a direct method, for identifying antigens. For the detection of antibody, a slide on which a preparation of the antigen on the slide is flooded with the specific serum, and, after an interval, the excess serum, is washed off.
The localisation of the antibody on the antigen preparation is then visualised under fluorescent microscope by layering the slide with the fluorescein labelled antibody specific for the serum proteins of the serum under test. The most suitable type of labelled antibody is that directed primarily against the globulin fractions of the test serum, i.e., fluorescein labelled anti-globulin serum.
(f) Antinuclear Factor Test:
Sera that are able to induce the formation of Lupus erythematosus (LE) cells contain the antinuclear antibody (ANP) which react with the nuclei of tissue cells. This factor present in the connective tissue disease (Serum Lupus Erythematosus, SLE) can be detected by the application of indirect fluorescent antibody technique.
(g) Antitoxin:
Toxin neutralisation fesris very useful to detect the toxigenicity of certain strains of bacteria, e.g., Clostridium tetani in mice. The control mouse, which has already been protected passively by Anti-tetanus serum (ATS) against tetanus, will survive, as the toxin is neutralized by anti-tetanus serum; the mouse which receives the toxin of toxigenic strain of CI. tetani, will show the symptoms and die of tetanus. The same principle is applicable in skin tests — Schick skin test to diagnose diphtheria and Dick test to diagnose scarlet fever.
(h) Anti-streptolysin-0 (ASO) titer:
Anti-streptolysin-O (ASO) titer is useful for the ‘ detection of clinically significant levels of Anti-streptolysin-O antibodies found in the serum of persons following infection with a streptolysin-O producing strain of streptococci. The ASO titer, expressed in Todd units, is the reciprocal of the serum dilution which completely neutralizes the streptolysin-O. A serum with a titer of 166 Todd units should show no hemolysis.
(i) Reverse Passive Hemagglutination (RPHA)test:
This test is a useful sensitive test to detect HBsAg in serum hepatitis patient. Besides, it is simple and specific.
A simple and convenient testing procedure to detect HBsAg:
25 µl diluent should be added into U or V cavity plates; then 7 μl test specimen (patient serum) should be added into the same cavity and mixed; 25 μl of sensitized sheep erythrocytes should be added and mixed again. The test should be kept at room temperature for 2 hours.
Interpretation:
Reactive screening test result – The cell settling pattern will appear as a large diffused pattern; non-reactive test result – a distinct compact bottom of cells.
(j) Enzyme Linked Immunosorbent Assay (ELISA) Technique:
In the micro-ELISA for detection of antibody, the antigen is coated onto the surface of wells in micro titration plate. The patient serum is then added and time is allowed for an antigen-antibody reaction to occur. An enzyme labelled specific anti-globulin is then added, which attaches to the antigen-antibody complexes.
Any untreated anti-globulin is washed away. An enzyme substrate is then added which is acted upon by the attached enzyme producing a colour which can be measured colorimetrically or interpreted visually. lncubation with enzyme substrate produces a yellow orange colour in the test well, if anti- HIV is present in the sample.
Commonly used enzymes are Horse Radish peroxidase (HRP) or phosphatase. Penicillinase prepared in India is also useful and cheaper than other enzymes. ELISA test is widely used to diagnose viral (AIDS, Hepatitis bacterial and parasitic diseases (Fig. 62.12).


(k) Western Blot Test:
Western Blot test is used to confirm HIV in patient sera screened positive for AIDS. This test is carried out only in AIDS Reference Laboratory (National Institute of Virology, Pune; AII India Institute of Medical Sciences, New Delhi; India).
Recent Parasitic Serology:
Serological tests are most helpful when the diagnostic forms cannot be readily demonstrated as in tissue infection such as amebic liver abscess (ALA), trichinosis, visceral larval migrans, toxoplasmosis. Immunodiagnostic tests to detect parasitic antigens have been described for a number of infections and could eventually replace morphological diagnosis of many parasites. Monoclonal antibodies may be particularly helpful in developing sensitive and specific antigen detection tests.
Antigenic structure of parasite is complex and cross-reactions in serological tests are common. Newer tests using different or more purified antigens have given better results. Various types of tests have been described. Agglutination tests use whole organisms or antigen coated particles such as in bentonite flocculation (BF), indirect hemagglutination (IHA) and latex agglutination (LA).
Complement fixation (CF) test has been widely used but is being superseded by newer tests. Precipitin tests include capillary precipitin, double gel diffusion, counterimmunoelectrophoresis (CIE) and circumlarval precipitin. Indirect immunofluorescence (IIF) has been widely used in parasitic serology and is readily adapted to detect Ig M antibody.
Recent test development has emphasised Enzyme linked immunosorbent assay (ELISA) and soluble antigen fluorescent antibody (SAFA) such as Automated fluoroimmuno assay (FIAX) system, although some improved tests are based on modification of older methods (e.g., IIF, CIE).Thus improvements in serological methods and antigens are causing rapid changes in parasitic serology.
Cross-reactions are common and sensitivity and/ or specificity of some tests maybe poor. In addition, serology may not distinguish between previous and active infection. Thus, for some infection, serology may be useful in screening person who have visited an endemic area, whereas they are of little value in diagnosing infection in residents of the endemic area. The presence of antibody does not necessarily indicate immunity. Immunodiagnostic procedures for detecting antigen or antibody in parasitic diseases are numerous and are changing rapidly.
ELISA Technique:
In the micro-ELISA for detection of antibody, the antigen is coated on to the surface of wells in micro titration plate. The patient serum is then added and time is allowed for an antigen-antibody reaction to occur. An enzyme labelled specific anti-globulin is then added, which attaches to the antigen-antibody complexes.
Any unattached anti-globulin is washed away. An enzyme substrate is then added which is acted upon by the attached enzyme producing a colour which can be measured colorimetrically or interpreted visually. Commonly used enzymes are peroxidase of phosphatase. Penicillinase is found useful, cheaper and can be prepared in India.
In recent years, ELISA is used to assist in the diagnosis of malaria, amebiasis, schistosomiasis, onchocerciasis, leishmaniasis, echinococcosis, trypanosomiasis, trichinellosis and toxoplasmosis. ELISA is sensitive and specific than other serological tests.
Amoebiasis:
Serodiagnostic procedure depends on the type of infection present, a very low degree of sensitivity is found with sera from asymptomatic carriers, increased sensitivity from patients with amebic dysentery and the greatest sensitivity with sera from those patients with extra-intestinal disease.
The CF has been generally replaced by IHA, CIF and indirect fluorescent antibody (IFA) procedures, these three tests have more or less the same degree of sensitivity. A new fluorescence (FIAX) technique — in which the fluorescence is measured in a fluorometer — is a new test that has been adapted to routine diagnosis of amebiasis. The technique has been developed for detection of antigen in feces. The sensitivity of this test is quite good, although the only parasite detected is E. histolytica.
Invasive amebiasis by E. histolytica can be detected by a very recent technique — cellulose acetate precipitation (CAP) test.
Dot immuno-binding assay (DIB) and sandwich ELISA can be used in the diagnosis of invasive amebiasis. Both tests are equally specific and sensitive. DIB, easier to perform is less expensive and recommended for detection of antibody in patients with invasive amebiasis (amebic liver abscess — ALA) in India (1992).
Giardiasis:
ELISA is compared with microscopy for detection of Giardia fecal antigens. ELISA was highly sensitive and specific either visually (95 and 97 per cent, respectively) or by optical density determination (99 and 96 per cent, respectively). ELISA is extremely effective for epidemiology study.
Toxoplasmosis:
The methylene blue dye (MBD) test — Sabin-Feldman dye test — has been used for many years for the serological diagnosis of toxoplasmosis. This procedure has been replaced by the IHA and IFA procedures, both of them are technically simple to perform and utilise a killed antigen rather than live organisms (used in MBD test).
All these procedures are equally sensitive and specific procedure can be performed using specific conjugates of Ig M — although the interpretations of the results are difficult. Congenital infections are generally indicated when sera from newborns are positive with Ig M conjugates.
Ig M antibody methods may give both false positive and false negative results, because of rheumatoid factor, blocking antibody and the induction of factor Ig M antibody against maternal globulin. The double sandwich ELISA test has eliminated many of these problems. The combination of IFA and IHA procedures allows more accurate interpretation, because a different type of antigen is used for each test.
Tests for Ig M antibody (Ig M fluorescent antibody or double sandwich ELISA technique) are particularly useful for establishing recent toxoplasma infection, because titers appear early (as early as five days after infection) and disappear within several months. Ig M antibodies are elevated in acute disease. CFT using soluble toxoplasma antigen becomes positive three to six weeks after infection, rises for two to eight months, and falls to a very low level after one to two years.
Very recently, the direct agglutination test with 2 mercaptoethanol (Ad-2ME)and immuno-fluorescent antibody test (IFAT) are used in the diagnosis of toxoplasmosis. The former is little superior to the latter.
Pneumocystosis:
The tests of choice for Pneumocystis are the CF and IFA tests and can detect 85 per cent infected patients. A direct fluorescent antibody test has been developed for the detection of organisms in mucus and sputum smears and tissue biopsies.
During the last few years, CIE and ELISA were evaluated. They are not still easily available. They lack specificity and sensitivity; so they are not used successfully in routine clinical diagnosis. The development of culture technique for this organism may lead to more specific antigen production.
Leishmaniasis:
The serological procedures (IHA, IFA and CF) are available for visceral leishmaniasis and are quite helpful in making diagnosis and IFA is being used routinely with excellent results using amastigote antigen for cutaneous leishmaniasis. The IFA test is more specific than ELISA for diagnosis of cutaneous leishmaniasis, but neither is satisfactory as microscopic examination. Both methods were acceptable for the diagnosis of visceral leishmaniasis when promastigote forms of L. donovanivuere used as antigen. ELISA test, using promastigotes as antigen, is probably the most practical for testing number of sera.
The IFA technique employing either amastigotes or promastigotes as antigen has been used as direct agglutination test (DAT) of fixed promastigotes. There may be cross-reaction with serum of patient infected with T.cruzi. The leishmanin skin (delayed hypersensitivity) test is negative in cases of active visceral leishmaniasis (AVL).
In 1995, an immuno-dot assay was developed for sero-diagnosis of AVL which utilizes protein. A colloidal gold as the visualizing agent. The test is simple requires few reagents and can be completed in 2 hours. It is sensitive and specific for AVL and generally correlates with ELISA. Either whole blood or sera in minute quantities may be used as test antibody.
Besides indirect ELISA tests for L. donovanianti-body was carried out with enzyme labelled protein A or protein G instead of anti-immunoglobulin. It was found that this modification improved the positive/ negative discrimination. In addition, antibody could be measured in human sera (e.g., dogs).
In 1995, monoclonal antibody L12 F7 against the antigen of L. donovani promastigotes was labelled with peroxidase and used in dot-ELISA test for detecting circulating antigen in sera of cases with visceral leishmaniasis. The results showed that monoclonal antibody can be used in dot-ELISA for diagnosis of visceral leishmaniasis. It is of special significance that this method can be used as simple and reliable tool for filed evaluation of therapeutic effectiveness for visceral leishmaniasis.
A technique that may permit direct and rapid diagnosis of cutaneous leishmaniasis as well as differentiation of leishmania is blotting with radiolabeled DNA probes. A positive leishmanin skin test and serum antibody can be demonstrated in patients by the time a cutaneous lesion has ulcerated. These tests remain positive in mucocutaneous disease. Serological tests are not useful in the diagnosis, because antibody levels are low. Positive skin test and serological test can reflect a previous rather than a current leishmanial infection. A direct agglutination test (DAT) is useful and sensitive in the diagnosis of cutaneous leishmaniasis.
Chagas Disease:
Serological testing is generally not needed for diagnosis of acute disease. Parasite- specific Ig M antibody detected by immunofluorescence or direct agglutination does not become positive until twenty to forty days after the onset of symptoms. The diagnosis of Chronic Chagas disease requires demonstration of antibody to T. Cruzi in the presence of characteristic cardiac abnormalities. In 1995, in Brazil, American trypanosomiasis (T. Cruzi) can be diagnosed by testing blood donor for infection by G-agglutination test (WHO).HIA and 11F tests are also used.
African Sleeping Sickness:
Several immunodiagnostic tests have been developed for African trypanosomiasis IFA, ELISA, Capillary hemagglutination (HA) test that are useful for epidemiological surveys. However, no serological test provides sufficient definite information for treatment of a patient without demonstration of the organism. A card agglutination test (CATT) has been produced in at el 996. It is simple, specific, sensitive. It is yet to be fully evaluated.
Malaria:
A variety of serological tests have been developed for malaria, but are not usually used for the diagnosis of clinical infection. They are particularly useful for epidemiological survey and detection of infected blood donors. Those most commonly used are indirect immuno-fluorescent (IIF) and IHA tests.
IIF titres equal to or greater than 1:64 are suggestive of recent infection with plasmodium. These serological tests showed a false positive rate of 1 per cent or less and have a sensitivity of over 95 per cent. Development of natural immunity in P.falciparum malaria can be detected by Western blot. The rather innovative title “ABC – ELISA” is merely a novel abbreviation for avidin-biotin complex ELISA.
It is useful method for malaria serology in the field and is most recent. A conventional ELISA is carried out but instead of enzyme labelled anti-globulin, biotin labelled anti-globulin followed by avidin labelled enzyme (1 additional step).The results of this new test correlated well with those of obtained by immunofluorescence and was better than those of the conventional ELISA.
The IFA test and micro-ELISA test for detection of antimalarial antibodies have little value in endemic areas. Recently, monoclonal antibody has been introduced for the detection of plasmodial antigen inhuman blood for the diagnosis of malaria by IFA technique.
In India, IIF test is used for the diagnosis of malaria by using P. falciparum antigen. The diagnostic titre is 1:80. Besides, 80 per cent seropositivity was observed by mean ELISA — optical density values to both P.falciparum and P. vivax. Plasmodial antigen can be detected by monoclonal antibody as a screening procedure for blood donors in transfusion medicine in epidemic countries like India.
Cryptosporidiosis:
A serological test has been described (J. Clin. Microbiol. 18:165, 1983).
Trichinellosis:
Diagnosis is often established indirectly by serological tests, some are commercially available. The most commonly used are bentonite flocculation (BF), fluorescent antibody (IIF) and Complement fixation (CF).The bentonite flocculation (BF) is very sensitive and usually becomes negative two to three years after an infection; thus, a positive test usually indicates active infection.
The test becomes positive after the third week of infection and is most helpful when a fourfold rise in paired sera can be demonstrated. The CF test detects antibodies slightly earlier than the BF test. IIF is considered to be as good as the BF test and most sensitive.
Filariasis:
Serological tests using extracts of Dirofilaria immitis as antigen include IHA and BF, but they allow only a diagnosis of filarial group rather than the species. Moreover, there is cross-reactivity with other parasite (false positive) as well as many false negative. Western blot technique and ELISA can be used to detect B. malayi in filarial sera of different groups of patients.
Schistosomiasis:
Several serological tests (Cholestrollecithin flocculation, BF, CF and IFA) are used for the diagnosis of schistosomiasis, all share problems of specificity and sensitivity. There has been increased use of IFA procedure which uses section of adult worm for the antigen and proved to be most sensitive technique (less cross reactivity with other sera). The circumoral precipitin test is used extensively for diagnosis of S. japonicum infection. ELISA technique is sensitive as the IFA and CF titers with adult worm using as antigen, but indicates that ELISA was more specific.
Cysticercosis:
IHA was 85 per cent reactive with sera from proved cases of human infection. The double diffusion was sensitive using both animal and human sera. Cross-reaction with sera from patients infected with Echinococcus species, T.saginata and Coenurus species have been reported-
Echinococcosis:
The IHA, IFA and immuno-electrophoresis (IE) procedures are considered to be the test of choice for the diagnosis of echinococcosis, IHA and BF are routinely used at Centre for Disease Control (CDC), USA, the IHA is most sensitive. Much IHA titers usually indicate the presence of hydatid disease.
IE test has been evaluated in many countries, a double diffusion band 5 (DD5) test has been reported to be more sensitive and more specific than IE test. CIE test has also been reported to be specific and sensitive and ELISA has also been evaluated. IHA titer of 1:256 is considered significant and is positive in 88 per cent of people with non- calcified hepatic cyst.
Low titer with any of these tests do not necessarily mean infection, since sera from patients with other conditions, such as liver cirrhosis and collagen diseases, cross-react. Cross reaction with the cestode larvae such as cysticercus are also common. In addition, patients with calcified hepatic cysts or cysts of lung frequently have negative serology.
Visceral larva migrans:
Serological tests may aid in establishing the diagnosis. Different methods vary in sensitivity and specificity. The ELISA using embryonated egg extract as antigen offers much better sensitivity and specificity and cross-reacting with Ascaris antibody can be removed by pre-adsorption. In visceral larva migrans, a significant titre is 1:321 whereas if ocular toxocariasis is suspected a titer of 1:8 is significant.
Strongyloidiasis:
The results of the indirect agglutination test with newly developed gelatin particles were found to be quite comparable to those of IHA and ELISA. The test is simple and rapid to perform for mass screening for human strongyloidiasis very recently, the sensitivity, specificity, positive and negative predicted values of ELISA test using F2, protein fraction from S. stercoralis were 95, 96.4,95 and 96.4 per cent, respectively, as the latest observation. Immunoblot can be useful in the diagnosis of human strongyloidiasis.
Filariasis:
In India, very recently, Dot-ELISA in detection of W. bancrofti filarial antibody was compared with standard ELISA, the dot-ELISA was found more sensitive. Besides, ELISA developed using soluble antigen of adult B. malayi gave positive responses in 95 per cent cases in India.
Onchocercosis can be diagnosed by recent immunoblot test:
Dracunculosis:
Most recently, the Falcon assay screening test — enzyme linked immunosorbent assay (FAST-ELISA) and enzyme linked immunoelectrotransfer blot (EITB) techniques are used to test human sera with D. medinensis adult worm antigen.
Following serological tests are performed at Centre for Disease Control (CDC), USA (1990):

IHA—Indirect hemagglutination;
CF—Complement fixation;
DAT—Direct agglutination test;
IIF — Indirect immunofluorescence;
BFT—Bentonite flocculation test;
ELISA —Enzyme Linked Immunosorbent Assay; + Any
IIF – Ig M titer to toxoplasmosis in an infant less than two years old is strongly suggestive of infection.
The diagnostic titres given are highly suggestive of clinical disease, but a fourfold rise in titer is stronger evidence.
Infections due to Toxoplasma Gondii and Trypanosoma Cruzi:
The advent of Polymerase Chain Reaction (PCR) has opened up the possibility of isolating DNA sequence from ancient samples from museum specimens and archeological findings. As majority of these molecules in these ancient samples are degraded to such an extent as to preclude any other molecular technique, PCR with its remarkable sensitivity can be employed to amplify the existing few molecules to an appreciable extent which can then be further studied.
For example, for the diagnosis of infections caused by RNA viruses by PCR, RNA has to be first converted into the complementary DNA (C DNA). C DNA is obtained by a reverse transcriptase assay using RNA as the template. This step takes 2 hours. The C DNA thus obtained is next used as the template in the PCR assay.
Serological tests which have wider use are commonly carried out and most of these tests are well-standardised. However, serological tests are essentially indirect tests except when antigens are detected. The more direct tests involve demonstration of the pathogen or pathogen specific nucleic acid (DNA or RNA) in the clinical diagnosis. Therefore, the Polymerase Chain Reaction offers a rapid, sensitive and a very specific diagnostic approach for protozoal diseases caused by Toxoplasma gondii or Trypanosoma cruzi.
Cultivation of Animal Viruses:
Viruses can be grown only within living host cells, as they gain access to the energy-producing systems and protein synthesizing machinery which they lack:
(a) Experimental animals,
(b) Chick embryos, and
(c) Tissue cultures.
(a) Animal inoculation was only old method available at that time by which the recognisable disease and death can be demonstrated. The presence of inclusion bodies in the tissues is the evidence of virus infections. The poliomyelitis virus can cause paralysis.
(b) Chick embryos (Fig. 62.13) provide more satisfactory hosts, because they are clean and bacteriologically sterile and have no specific protective immune system to counteract the virus infections. Poxviruses, vaccine, herpes virus can grow on chorioallantoic membrane (CAM, Fig. 62.13) and produce characteristic visible lesions. Influenza viruses multiply in the lung cells of the embryo and the cells lining allantoic cavity. Rickettsiae and psittacosis lymph granuloma group grow well in yolk sac and kill the embryo.

(c) Tissue cultures of human or simian cells are the most frequently used and are relatively simple to prepare in large quantities that are required by modern diagnostic virological methods. Suspensions of cells dispersed from tissue fragments or bulk cultures of cell lines (human amnion cell; monkey kidney cell; squamous cell carcinoma of the cervix uteri – He La cells; and HEP2 strain of from an epithelioma) are obtained by tryptin digestion. The cells adhere to the walls of the test tube and grow out to form a sheet — monolayer, which can be observed in situ under the low power objective of the microscope.
After inoculation into monolayer cell culture, a virus causes degeneration of the cell sheet. This progress of the infection (cytopathic effect CPE) can be studied under the low power objective of a light microscope and the cells may become rounded, shriveled, ballooned or fused together to form a multinucleate syncytial masses, ultimately all cells are affected and get detached from the wall of the test tube.
Cultivation of Fungi:
Cultivation of fungi can be done on Sabouraud’s medium at room temperature. The growth characteristic of each fungus can be studied along with its morphology, after staining with Lacto phenol blue, under low or high power objectives of light microscope.
Study of Blood Cell (Hematology):
Human blood cells are of three types:
1. Erythrocytes (red blood cells);
2. Leucocytes (white blood cells);
3. Thrombocytes (blood platelets).
In man, red blood cells (RBC) are round, devoid of nucleus, smaller than white blood cells. The cytoplasm of red blood cells contains a respiratory pigment called hemoglobin which imparts red colour. Due to hemoglobin, red blood cells help in transportation of respiratory gases, i.e., O2 and CO2.
White blood cells (WBC) are nucleated, colourless, irregular in outline and devoid of respiratory pigment. The cytoplasm of most of the white blood cells contains some granules and accordingly they are classified into two main groups: granular and agranular leucocytes.
According to the nature of these granules, granular leucocytes are further classified into three types:
(a) Neutrophils;
(b) Acidophils (Eosinophils) and
(c) Basophils.
(a) Neutrophils:
Granules of neutrophils as the name suggests, show neutral property. Their nuclei consist of three to five lobes. Sixty to seventy per cent of total leucocytes are of this type. They act as phagocytes and are also known as polymorphs.
(b) Acidophils :
Granules of acidophilsare alkaline in nature and are stained by acidic stains. Their nuclei are bilobed. Their percentage is very little, i.e., about 1 to 4 per cent of total leucocytes. They are also called as eosinophil’s, (c) Basophils are acidic in nature and stained by alkaline or basic stains. Their nuclei consist of three to five lobes. Their percentage is the least, i.e., 0 to 1.0 per cent of total leucocytes.
Agranular leucocytes are of two types:
(I) Monocytes, and
(II) Lymphocytes.
Monocytes are largest leucocytes, while lymphocytes are smallest. The nucleus of monocyte is crescentic, while that of lymphocytes is round. The number of monocytes is about 2 to 6 per cent, while that of lymphocytes is about 25 per cent of total leucocytes.
Lymphocytes, being smallest in size, can pass through the very thin wall of blood capillaries and, therefore, they are found in lymph. Hence, they are called as lymphocytes. Thrombocytes are small, oval or spindle shaped fragments of special large cells found in the bone marrow. Therefore they are devoid of nucleus and short lived. They disintegrate easily when they come out of the bleeding wound and help in clotting.
Hemoglobin (Hb) Report:
Blood counts Peripheral venous blood.
Hb.gms% norma/(foradults)
(Male, 13.5 to 18 g%; Female, 12 to 16 g%)
Total RBC /cmm (M,4.6 to 6.2 millions/cmm;
F,4.2 to 5.4 millions/cmm.)
Total WBC /cmm (4000 to 10,000/cmm).
Differential Leucocyte Count (DLC):
Erythrocytes Sedimentation Rate:
Bleeding and Clotting Time:
Bleeding time Up to 5 minutes.
Clotting time Up to 9 minutes (Lee & White method).
Blood Indices:
PCV% (42 to 60%)
MCV cmm (82 to 90 cmm).
Blood Examination for Parasites:
The blood provides the most common medium for recovery of human parasites at various stages. From this source, routine diagnosis is made of malaria, African trypanosomiasis, most types of filariasis, less frequently Chagas’ disease and rarely Kala azar and toxoplasmosis.
Preparation of Films:
Thin film on one slide. A drop of blood is taken at the end of the slide. A spreader is held at an angle of 45°C and pushed in the opposite direction to make a smear. Thick film on one slide. Four drops of blood are placed at the corners of a half inch square slide and joined to form a thick film. A glass slide with corners cut at one end or the coverslip of a hemocytometer can be used as spreader.
Examination of Blood for Malarial Parasites:
Time for taking blood. The blood specimen should be obtained several hours after the paroxysm has reached its peak, because the parasites are more easily detected in the film. The schizogony of Plasmodium vivax, P.malariae and P. ovale can readily be demonstrated in the peripheral blood during both the febrile and afebrile periods.
The best time for demonstrating P. falciparum is few hours after the febrile paroxysm reaches its peak, because P. falciparum disappears from the peripheral blood during the afebrile period. Generally, the blood film should be taken when the patient is seen first and therefore. It will be useful to have both thin and thick films, either on one slide or on two different slides and to examine them after staining. The thin film is examined to identify the parasites.
Stains:
1 .Leishman’s Stain:
Leishman’s stain should be poured on the smear and diluted twice its volume with neutral distilled water. The diluted stain is allowed to remain on the slide for 10-15 minutes, and then the slide should be washed with running tap water, dried and examined under an oil immersion lens.
2. Giemsa Stain:
The diluted Giemsa stain is poured over the film and kept for 30 to 45 minutes. The film is washed with running water and dried. The stain is examined under an oil immersion lens.
Examination of Blood for Microfilariae:
Blood should be collected between 10 p.m. an 2 a.m. for microfilariae are of nocturnal periodicity. A thick film of blood is prepared and kept covered. The next morning, it is dehemoglobinised by the slide being put in water, dried and fixed in methyl alcohol and then stained with Leishman’s stain or Giemsa stain. Microfilaria may be detected occasionally even in a thin film.
Stool Examination for Parasites:
The entire stool specimen should be examined macroscopically for its consistency and component elements (feces, mucus, blood tissue elements and undigested food) and for parasitic segments of Taenia saginata, T. solium, whole adult worm of Ascaris lumbricoides, Ankylostoma duodenale, Trichuris trichiura, Enterobius vermicularis and various intestinal flukes.
During the identification of eggs, one should pay attention to shape, size, colour and marking on the surface of egg shell, the presence of granules, ovum or a differentiated embryo, the existence of an operculum and the three pairs of embryonic hooklets in cestodes. For detection of helminthic eggs, only an unstained preparation is necessary (Fig. 62.14a, b).


Methods of Examinations of Stool, and in detecting parasitic infection is as follows:

1. Direct method is commonly used. Two cover glass preparations are prepared on one microscopic slide for apart from each other.
1. Saline preparation. 1 -2 mg. of the fecal material should be mixed in a drop or two of physiological salt solution for the left hand preparation and
2. Iodine preparation. 1-2 drops of iodine solution should be mixed with 1 -2 mg. of the fecal material for right hand preparation. Cyst forms and living parasites can be detected in the saline preparation. Nucleus, glycogen mass of the protozoan cyst of E. histolytica and the flagella on the parasite can be studied in iodine preparation.
Trichrome stain Kit with 1VA fixation was developed for the simplified staining of intestinal parasites. The cytoplasm of these organisms, cysts or trophozoites stain blue green with a purplish tint. The nuclear chromatin, chromatoid bodies, ingested RBCs and bacteria stain red or purple red. A pseudo-parasite is an object which resembles a parasite or egg of a parasite. A wide variety of objects which resemble parasites may be found in stool specimens.
Examination of Skin Scrapings for Identification of Mites:
A small quantity of skin scraping is taken in a test tube to which 10 per cent potassium hydroxide is added. The material is boiled for 10 minutes and kept in a stand to settle the sediment. A drop of sediment should be taken on a slide with the help of a pipette, covered with a cover slip and examined under low and high power objectives.
Examination of Urine:
Urinary tract infection can be confirmed by routine microscopy by presence of pus cells and by culture of urine or preferably centrifuged deposit on the laboratory media for the characteristic growth of organisms and the ultimate morphological identification of organisms.
Application to Nursing:
Though the nurse may not be well-acquainted with the handling of the microscope, staining and other laboratory techniques, she will be able to recognise the difficulties of the laboratory procedures and at the same time she will be able to differentiate Gram-negative and Gram-positive bacteria and the antibiotics effective on these broad groups of bacteria and the ultimate selection of an antibiotic for the treatment.
If a knowledgeable and professional nurse can recognise the problems and difficulties faced in the laboratory, she will be able to collect and handle the specimens, so that these specimens can be processed easily in the laboratory for the correct results. The same methods of handling of sterile materials in the hospital wards and in public health nursing by the nurse should be followed in the microbiology laboratory to avoid the contamination.
If the nurse has a sound knowledge of interpretation of the report of serology, blood examination for parasites, stool and skin scraping examination, she will be of great assistance in reporting immediately to physicians for the diagnosis and the appropriate treatment.